Using bcftools: utilities for variant calling and VCF files manipulation
Table of contents
Introduction
Background
There many variant callers, and the variants discovered differe to a certain extend between tools.
This is due to several reasons:
data preparation:
sequencing depth / coverage and quality
accuracy and or variation during read mapping (eg. different algorithms can lead to different optimal mapping) to a reference genome[1]
different algorithms and criteria during variant calling and filtering
You can read about some studies highlighting those variant calling differences. Some and non exhaustive examples: Hwang etal. 2015, Barbitoff etal. 2022, Andreu-Sánchez etal. 2021 , with recommendation for metagenomic samples, Mey Seah etal. 2023 and Olson etal 2015 for microbial genomics.
Taking control of the variant calling process and how variant files are manipulated can give you control of the whole process. It can help understanding what complex tools and pipelines actually do. Here we will give some examples on how you can do so with bcftools.
bcftools program is part of Samtools package suite. Bcftools is for example used in Snippy the variant calling and core genome alignment sowftware that is implemented in ALPPACA pipeline[2]. Snippy do not use bcftools for variant calling[3], but it uses it for several purposes: filtering variants, creating consensus, converting, compressing and indexing variant files. Bcftools offers a variety of commands/modules to manipulate VCF files. Combining those in different way, can help you extract the information you want from your data.
Examples of what can you do with bcftools and Samtools
Some examples of what you can do:
Call a consensus (using a reference and a variant call)
Variant calling, which includes:
choosing between two different variant calling models :
consensus-callermodel andmultiallelic-callermodeldetermining the likelihood of a genotype (understand of a variant in the pipelup), accounting for base and mapping quality
filtering variants by different criteria (eg. only calling variants that have been found in 90% of the reads of the pileup)
Calculating copy number variation (CNV) calling
Help ensure quality of your dataset by providing utilities to help detect sample concordance, swaps and contamination
Detect the number of chromosomal copies (polysomy)
It can also work with haplotypes, determine consequences of variations (eg Frame shift), and in association to specific mutation annotations (eg. as with SnpEff as done in Snippy) it can eg. help determine the effects of a mutation.
It also provide some utilities for files manipulations, and can provide statistics on variant calls, and much more. It seems eg. that it can help determining Hq variants by allowing to extract variants congruent between different variants callers (intersection).
Ressources
There are many resources online:
BCFtools HowTo, with manual and more generally Samtools website. A list of articles describing the algorithms and usages of bcftools is provided in Samtools documentation.
Videos on YouTube, eg. from Bioinformatics coach channel
Other tutorials that are relevant:
Galaxy training - Microbial variant calling with Snippy. We will use the data provided in this tutorial for the exercises.
You might have to consult the description of file formats. The standards for the following file formats can be found here.
Sequence Alignment Map format (SAM)
Variant Call Format (VCF)
The BED format
Terms to become familar with
Computer Science:
Binary VS non-binary variant files: BCF VS VCF - bcf is a specific form of compressed vcf. It is compressed with bgzip.
Working on stream - this means that one program produces an output that is then put as an input to the next without storing it as a file.
Indexing - this is creating a lookup “cheat table” for where things are in the file.
Note that Some operations only work on indexed files in bcftools, and Working with multiple VCF files will require indexing.
Standard vs non standard indexes
An important thing to remember is that when we are working with these tools, both the reference genome and the variant file can and should be indexed. The reference genome is indexed using samtools, while the variant file is indexed using bcftools.
bcftools / variant manipulation:
line interesections
phasing
mapQ : The mapping quality - as provided by samtools. Samtools mapping quality is computed as such:
−10 log10 Probability {mapping position is wrong}(phred correspondance)BAQ: per base alignment quality - Offset to base alignment quality - realignment
Be-aware that different aligners used mapping quality in a different way. You can read more in this blog and here. If you use other mapping tools, you might consider how the mapQ is used to adjust parameters accordingly.
Tutorial
In this tutorial we will first do variant calling using bcftools, and then we will explore the resulting files a bit.
Data used for this tutorial
We followed the Galaxy training - Microbial variant calling tutorial to provide a working data set you can exercise with. (Feel free to do the same). In this tutorial, Snippy is used to call variants between a read set and a reference genome. In this process, the reads are mapped to the reference, and the variants are subsequently called with freebayes. In this process, bcftools is used for manipulating files quite a bit.
In this tutorial we will recreate some steps used by Snippy to learn how bcftools works. This will also help us to better understand Snippy. We will eg. compare files generated with bcftools manually to what was generated during the Snippy run on galaxy.
The raw data and results from galaxy processing with Snippy for this tutorial is available in SAGA in:
/cluster/projects/nn9305k/tutorial/20240226_bcftools/data
And as you can see, we recovered the same variant position as in the galaxy tutorial:

bcftools in SAGA
Activate conda.
If you havent had set conda, please look at Thomas tutorial
miniconda
conda activate bcftools
# Version: 1.19 (using htslib 1.19.1)
bcftools --help
# OR
bcftools --version
Note: The most recent version of VCF tools is not compatible with
samtools <= 0.1.19. If you need to use VCF file produced by samtool prior to this version, you will need to convert to a more recent format.
To see the list of commands and plugins (aka what you can do with bcf tools do:)
# give you the list of default commands
bcftools
# give you an overview of additional pluggins
bcftools plugin -lv
Note on bcftools usage
It is possible that you will have to compress input files to be able to work with bcftools. We will see an example on how to do so during consensus calling.
Variant calling
Variant calling requires two phases:
The first step is the computation of the likelihood of each genotype (understand the likelihood that each detected variant is true). This is done with
bcftools mpileup. This tool will look at the bases in the bam file that have been “stacked” up on each position, and calculate the likelihood of what base should be in each position. The result file will contain both variants and non-variants.The second step is the actuall call of variants. This is done with
bcftools call. This is a filtered and edited version of what comes out of the mpileup program, focusing on where the variants are.
To start the process of variant calling, you need an indexed reference. If the reference is not indexed yet, you need to create an index. This is done with samtools:
Note: usually you already have an indexed reference from the mapping step.
samtools faidx ../wildtype.fna
Get options used for computation of likelihood:
bcftools mplieup
mplieup options you might want to consider adjusting:
-d OR --max-depth <YourDepth>Per default the maximum depth of the pileup that is used is 250. If you have sequenced deeper, adjust this.-q, --min-MQ INTwhich allows to filter out alignments that have a quality (mapQ) lower than a certain threshold-Q, --min-BQ INTto avoid accounting for bases with a quality lower than a certain threshold-rlist OR-Rfile to select certain regions of the reference only. This can eg. allow you to avoid calling variants in repeated regions.There are options to ajust how the likelihood is computed (we keep those for now as we have little experience, per default Illumina profile is activated)
--seed INTallows you to set the seed - this can be usefull if you try to adjust options
Options used here:
Ououtput type uncompressedfformat of fields. To do specific format for the columns of VCF file (see VCF standard FORMAT column)
Note that at this stage the likelihood is computed assuming a diploid organism. You will have to adjust the filter to organism ploidy during variant calling.
Get options used for variant calling:
bcftools call
call options you might want to consider:
-rlist OR-Rfile to select certain regions of the reference onlyoptions to adjust usage of the likelihood that was calculated
used here:
Ozoutput type compressed (bgzf) orOuoutput uncompressedooutput file name-cOR-mis the variant calling model chosen (pick one).voutput variant sites only (this is what is a VCF)--ploidy
Note:
-cconsensus-caller model is the originally developped model, while-m, the multiallelic-caller which is used for rare-variants and multiallelic calling is NOW the default method used in bcftools.
Example for the tutorial:
cd tutorial
# bcftools mpileup -Ou -f <ref.fasta> <mapped_reads.bam> "the result of this is piped to:"
# bcftools call -mv -Oz -o <called_variants>.vcf.gz
bcftools mpileup -Ou --max-depth 500 -f ../wildtype.fna ../galaxy_snippy/snippy.bam -Ou -o result_mpileup.vcf
bcftools call -mv -Ou --ploidy 1 -o calls.vcf result_mpileup.vcf
# OR in one command
bcftools mpileup -Ou --max-depth 500 -f ../wildtype.fna ../galaxy_snippy/snippy.bam | bcftools call -mv -Ou --ploidy 1 -o calls.vcf
Have a look at both the vcf files that are produced here. To make it easier, use ls -S filename. As you can see you have a lot of things in the file produced first, and much less in the second.
Find which VCF file format is used and look at the corresponding format standard
What does Snippy uses bcftools for ?
You can have a look at the commands used by Snippy
in the tutorial data/cluster/projects/nn9305k/tutorial/20240226_bcftools/ subfolder data/galaxy_snippy/snippy.txt file.
Consensus calling
Snippy uses bcftools[5] to in some ways “merge” the reference and the variants. It basically puts the variants on top of the reference and outputs that. Sometimes this is called a pseudogenome.
bcftools consensus --sample mutant_R1_fastq -f reference/ref.fa -o snps.consensus.fa snps.vcf.gz
-ois the consensus output file - fasta format
snps.vcf.gz is the compressed form of the final and annotated variant file
ref.fais the wild strain reference (it only differs by encoding in upper vs lower cases)
mutant_R1_fastq is the sample name
We create a copy of the VCF file and reference in a separate directory to be able to follow our work
We need to index and compress the VCF file (bgzip). The compressing utility is installed with samtools/bctools
We recreate the consensus
We compare our consensus to Snippy consensus. Because in theory those sequences should be identical (obtained by the same means, but it might have been some headers reformating), we can concatenate “our” consensus and Snippy consensus and pretend this is an alignment and detect SNPs differences between those consensus files.
Note: We used the final files provided by galaxy to demonstrate equivalence
# 1. Create dir, copy of the VCF and ref files
cd data && mkdir consensus && cd consensus
cp ../galaxy_snippy/snippy.vcf .
cp ../wildtype.fna .
ls
# 2. Compress and index the VCF file
conda activate bcftools
bgzip -ik snippy.vcf
# there is a problem with the index that is given as output (bug) we fix that
bcftools index snippy.vcf.gz
# Creating the consensus
bcftools consensus --samples mutant_R1_fastq --fasta-ref wildtype.fna --output our_consensus.fasta snippy.vcf.gz
conda deactivate
# Comparison to snippy consensus
# In theory those sequences should be identical (except maybe header)
# create a dummy alignment
cd ..
cat galaxy_snippy/snippy_consensus.fasta consensus/our_consensus.fasta > dummy.fasta
conda activate snp-dists
snp-dists -a dummy.fasta # no differences were detected
conda deactivate
Variant filtering
Why filter variants, an example of variants being filtered out because of lack of depth in the pileup.
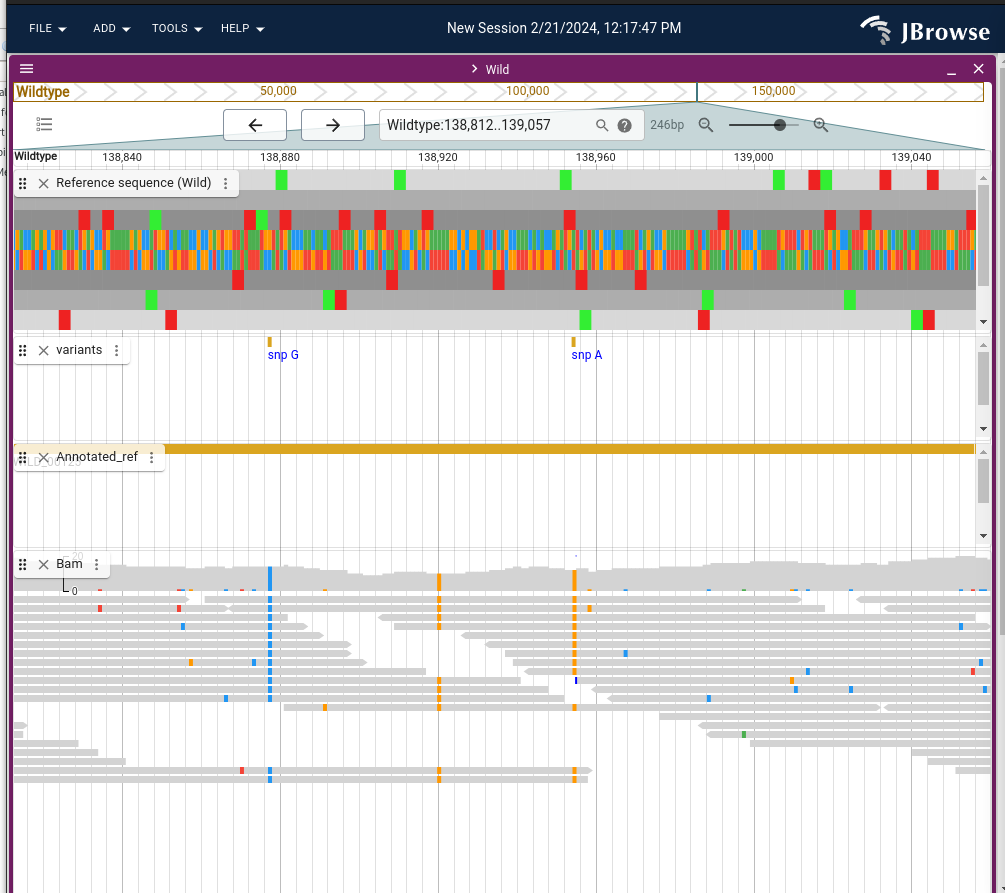
The unfiltered variants called with FreeBayes when running Snippy can be found here:
data/galaxy_snippy/snippy_zip/mutant_R1_fastq/snps.raw.vcf
and the filtered variant call here data/galaxy_snippy/snippy_zip/mutant_R1_fastq/snps.filt.vcf.
Note: The final annotated variant[4] are provided in data/galaxy_snippy/snippy_zip/mutant_R1_fastq/snps.vcf.
Note: This last one is equivalent to the one exported as
data/galaxy_snippy/snippy.vcf. You can test this usingdiff file1 file 2
cd data
diff galaxy_snippy/snippy_zip/mutant_R1_fastq/snps.vcf galaxy_snippy/snippy.vcf
We need to understand also to look at the unfiltered variant and find which VCF format was used, and gain information about the meaning of the fields. We need also to compare this file with the filtered variant file, and understand what is done in Snippy.
cd data/snippy/mutant_R1_fastq
less snps.raw.vcf
Which VCF version was used ?
The VCF file standard used is VCFv4.2. Bellow an extract of the relevant fields.
Which reference was used, its ID and length
You can see the command that was called to call FreeBayes.
That no phasing information was used.
The fields descriptions for the columns provided in the VCF file
Description of metadata content in the VCF file
INFO : description of the fields
FORMAT/TAG = FMT/TAG <!– TAG is the flag used to describe what type of info is contained>
FILTER : Description of the filters that have been applied to the data
VCF fields
CHROM : contig / chromosome of the reference
POS : position according to the reference (base 1)
ID : type of structural variant <!– no structural variant so . >
REF : the nucleotide in the reference
ALT : the alternate nucleotide(s) in the sample (comma separated)
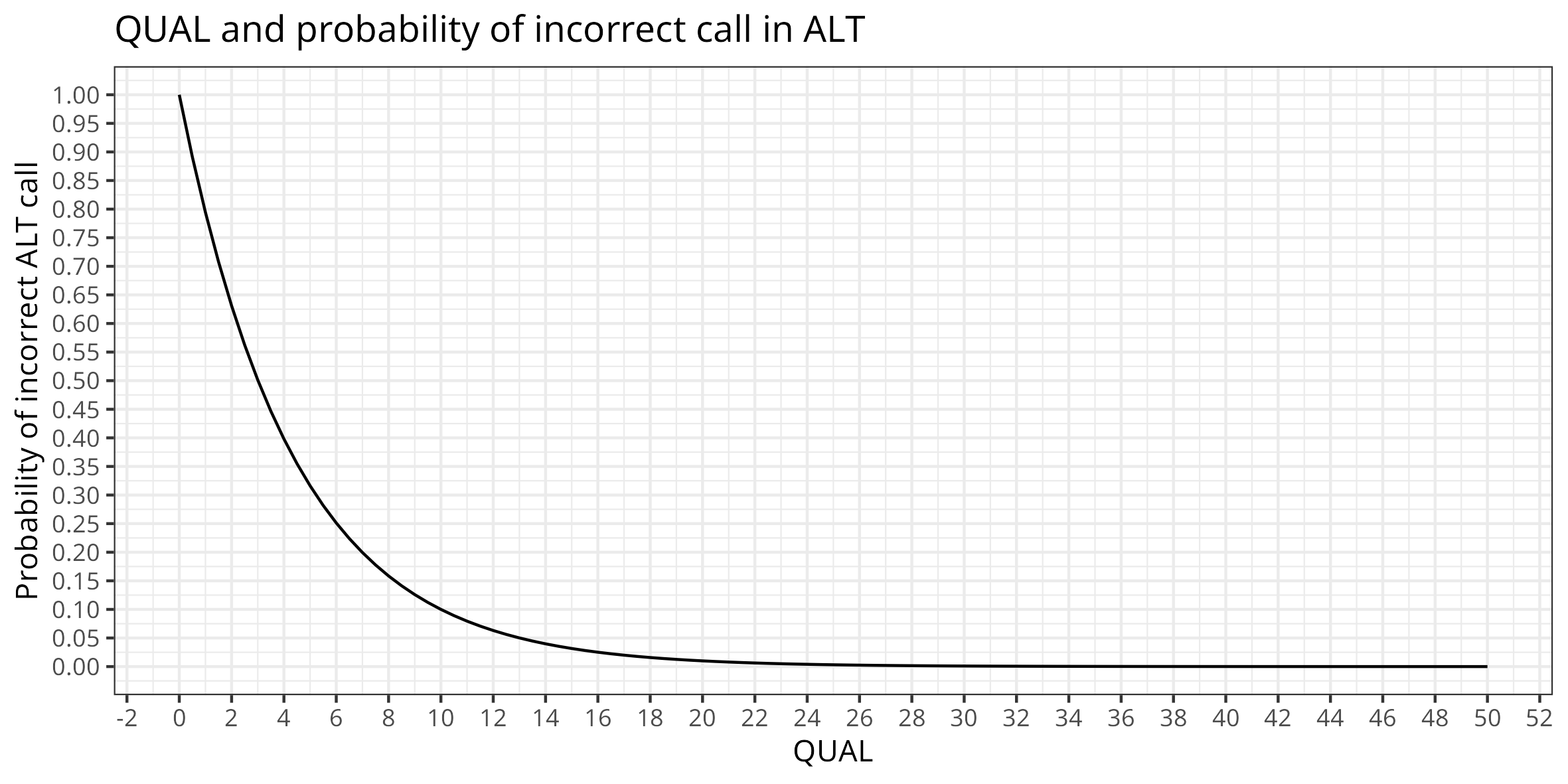
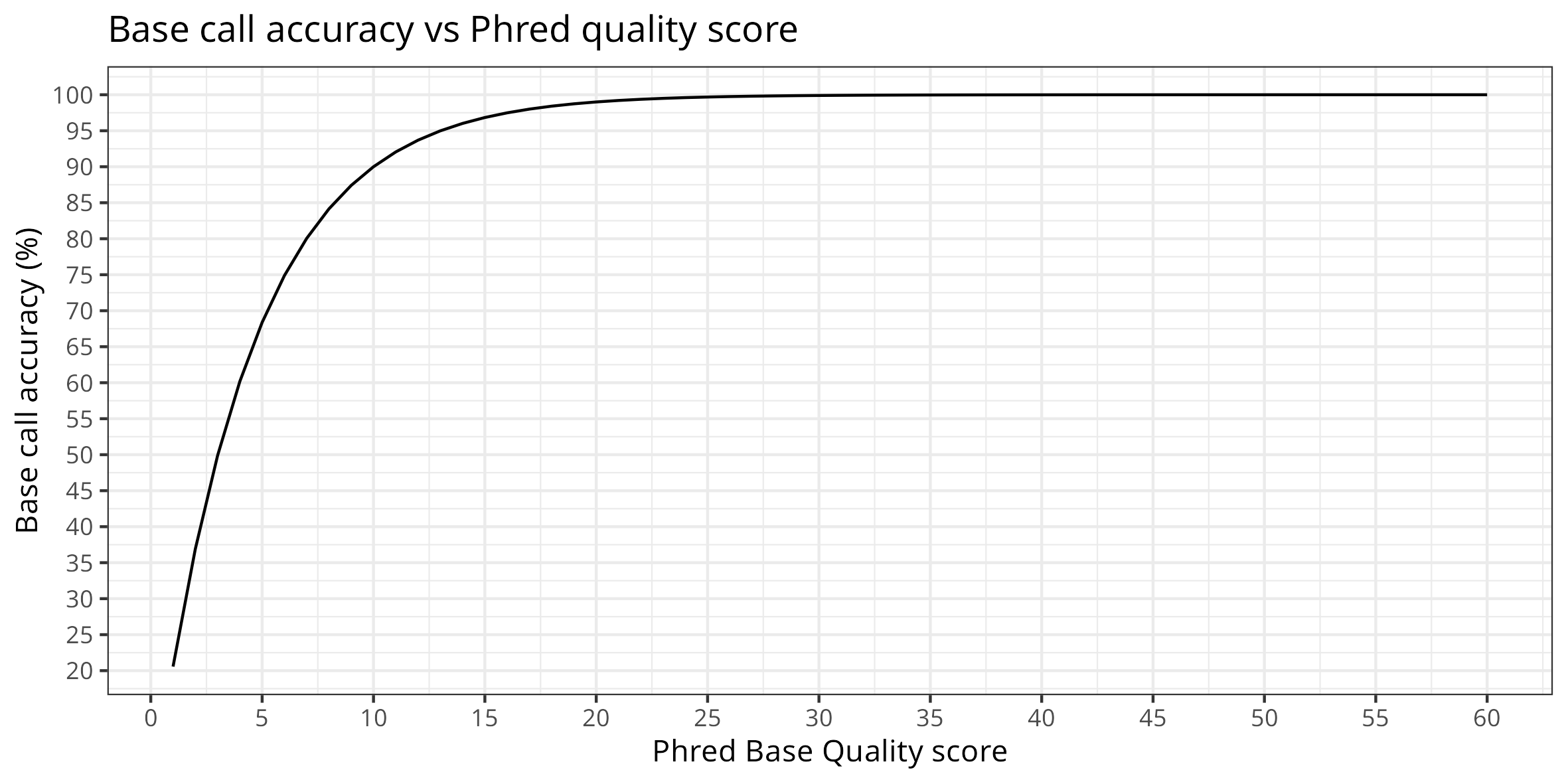
QUAL : Phred-scaled quality score for the assertion made in ALT. “−10log10 prob(call in ALT is wrong)”.
FILTER : filter status : PASS if go through all the filters, otherwise list of filters where it failed <!– no filter>
INFO: Additional information for the variants detected (see descriptions of tags). We can look at some of those
AC: allele count in genotypes
AF: allele frequency (in order allele is listed)
AN: total number of alleles in called alleles
DP: combined depth across samples at the position (eg. here we used one sample so it gives the depth)
LEN: the allele length
TYPE: describe the type of variant (snp, del, ins, mnp: several bases changes) …
Some information can be redundant with format
FORMAT : read specific to vcf file you have, some abbreviations may change
GT : genotype encoded (diploid ./. haploid . - / means unphased otherwise would be | )
DP : here its the read depth
AD : number of observations for each allele
RO : reference allele observation count
QR : Sum of quality of the reference observations
QA : Sum of quality of the alternate observations
AO : Alternate allele observation count”
GL : genotype likelihod (ref, diploid - 1 for each allele)
Looking at Snippy very long filtering command …
Note that snippy uses an earlier version of bcftools, this is why the command is sligly different than the command used in our tutorial
bcftools view --include 'FMT/GT="1/1" && QUAL>=100.0 && FMT/DP>=10 && (FMT/AO)/(FMT/DP)>=0.9' snps.raw.vcf | vt normalize -r reference/ref.fa - | bcftools annotate --remove '^INFO/TYPE,^INFO/DP,^INFO/RO,^INFO/AO,^INFO/AB,^FORMAT/GT,^FORMAT/DP,^FORMAT/RO,^FORMAT/AO,^FORMAT/QR,^FORMAT/QA,^FORMAT/GL' > snps.filt.vcf
Lets decompose : 1 -> 2 -> 3 -> 4
bcftools view --include 'FMT/GT="1/1" && QUAL>=100.0 && FMT/DP>=10 && (FMT/AO)/(FMT/DP)>=0.9' snps.raw.vcfviewallows to view, filter and subset VCFGT=1/1 Genotype must be identical (homozygosity) : here is the transfromation from the diploid encoding so it gets valid with halpoids
QUAL>=100 guet as close as possible as 0 probability incorect ALT call (adjust not to filter all!)
FMT/DP>=10 : Filtering if depth at locus is < 10
(FMT/AO)/(FMT/DP)>=0.9 - AC >= 0.9 read depth to be acounted for
vt normalize -r reference/ref.fa- Snippy use vt tool to do an operation called variant normalisation, which standardize the way variants are represented in the VCF file.
bcftools annotate --remove '^INFO/TYPE,^INFO/DP,^INFO/RO,^INFO/AO,^INFO/AB,^FORMAT/GT,^FORMAT/DP,^FORMAT/RO,^FORMAT/AO,^FORMAT/QR,^FORMAT/QA,^FORMAT/GL'annotateallows to add or remove annotation to VCF files. So its a way to slighly clean the long VCF file from the fields the author does not want to keep.
output the filtered variants to file:
> snps.filt.vcf
Can we reproduce this type of filtering?
# copy input in
mkdir filter_variants && cf filter_variants
bcftools view --include 'FMT/GT="1/1" && QUAL>=100.0 && FMT/DP>=10 && (FMT/AO)/(FMT/DP)>=0.9' -Ou -o filter_step1.vcf snps.raw.vcf
Line in VCF are added to show the filtering column, and we can crosscheck that the variants are filtered according to those criteria, we can see in the vcf file that the loci that did not fullfill criteria are filtered out
The normalisation of bcftools appear to be slighly different than the one from vt tool. bcftools seems to only left align, whereas vt tool appear to apply an additional parsiomny criteria.
There are also options to split or combine multiallelic sites (VCF manipulation). To do a simple left alignment, one can do:
bcftools norm -f wildtype.fna -Ou -o filter_step2.vcf filter_step1.vcf
And then we can filter the fields in the vcf file
bcftools annotate --remove '^INFO/TYPE,^INFO/DP,^INFO/RO,^INFO/AO,^INFO/AB,^FORMAT/GT,^FORMAT/DP,^FORMAT/RO,^FORMAT/AO,^FORMAT/QR,^FORMAT/QA,^FORMAT/GL' -Ou -o filter_step3.vcf filter_step2.vcf
This also add the last command to the VCF file.
Check difference between filtered variant file and our results. The same variants remain. However we do not have the effect annotations yet.